
CARDIOMYOPATHIE DILATÉE
Brochure patients
Document rédigé par l’équipe pluridisciplinaire du centre de référence pour les maladies cardiaques héréditaires (Paris), en collaboration avec des patients atteints de la maladie
Elle appartient au groupe des Cardiomyopathies, qui désignent des maladies du muscle cardiaque (myocarde) sans cause apparente. La Cardiomyopathie Dilatée est caractérisée à la fois par une dilatation des ventricules (c’est-à-dire une augmentation de taille des cavités cardiaques, alors que les parois musculaires restent minces) et par une hypokinésie de ces ventricules (c’est-à-dire une faiblesse de la contraction du muscle cardiaque). Elle concerne de façon prédominante le ventricule gauche, mais le ventricule droit est fréquemment impliqué.
Le diagnostic de Cardiomyopathie Dilatée (CMD) suppose d’avoir éliminer les autres causes de cœur dilaté et hypokinétique, telles que l’infarctus du myocarde ou les maladies de valves cardiaques.
La maladie conduit à une altération de la pompe cardiaque, avec un débit sanguin qui peut devenir insuffisant pour assurer les besoins de l’organisme. La principale manifestation est alors l’essoufflement à l’effort, lié à une accumulation sanguine en amont du cœur gauche avec congestion des poumons. La diminution de la perfusion des reins peut entraîner une mauvaise excrétion de l’eau et du sel, aboutissant à une rétention et à des oedèmes (notamment au niveau des jambes). L’accumulation sanguine en amont du cœur droit y participe aussi. L’ensemble de ces symptômes correspond à l’insuffisance cardiaque.
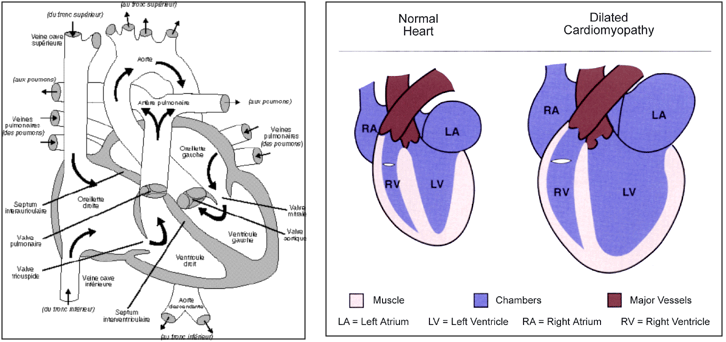
Le cœur normal (à gauche) et la cardiomyopathie dilatée (à droite)
La maladie est considérée comme assez rare, avec une fréquence d’environ une personne sur 3000 dans la population générale. La fréquence réelle de la maladie pourrait être très supérieure à cette estimation.
Par ailleurs la maladie peut se rencontrer à tout âge mais préférentiellement chez l’adolescent et le jeune adulte. Parfois, certains patients évoluent vers une insuffisance cardiaque sévère, qui peut conduire à une transplantation cardiaque.
Lorsque l’on considère l’ensemble des maladies cardiaques qui conduisent à une greffe cardiaque (souvent proposée chez des sujets jeunes), la Cardiomyopathie Dilatée en constitue l’une des principales indications.
La Cardiomyopathie Dilatée n’est pas toujours génétique. Elle est parfois liée à une infection virale du myocarde, parfois liée à une réaction immunitaire anormale (dite auto-immune), parfois liée à une consommation excessive d’alcool (avec régression ou guérison possible après sevrage et abstinence), parfois liée à une grossesse (Cardiomyopathie dite du péripartum). Parfois, la cause est génétique (dans 20 à 35% des cas de CMD), avec un contexte familial alors souvent présent.
Lorsque la maladie est d’origine génétique la transmission est souvent autosomique dominante, ce qui signifie que le gène anormal peut se transmettre à la descendance avec un risque de 50% pour chaque enfant, et un risque équivalent pour les garçons et les filles. Dans certains cas cependant, la maladie se transmet selon un autre mode : autosomique récessif, lié au chromosome X, ou encore selon une transmission mitochondriale.
Plusieurs gènes sont impliqués dans la maladie (plus d’une quinzaine), avec diverses anomalies (mutations) identifiés dans ces gènes (par exemple dans le gène codant la chaîne lourde bêta de la myosine ou MYH7). La découverte de ces anomalies a constitué une étape importante, mais le mécanisme précis de la maladie est encore obscure et la compréhension de la maladie est très incomplète. De plus les gènes actuellement connus ne sont en cause que dans une petite partie des familles. Les études de recherche se poursuivent pour identifier de nouveaux gènes, et pour mieux comprendre le mécanisme de la maladie.
Dans les formes génétiques de la Cardiomyopathie, les anomalies cardiaques (ventricule dilaté et hypokinétique) sont rarement présentes dès la naissance. Elles apparaissent et se développent secondairement, chez l’enfant, l’adolescent, parfois bien plus tardivement chez l’adulte.
Certains gènes sont responsables de formes cliniques particulières. C’est le cas des « Laminopathies », dues à des mutations du gène des lamines A/C (ou LMNA). Cette maladie peut se manifester par (i) des troubles de la conduction électrique (bloc auriculo-ventriculaire ou dysfonction sinusale, pouvant conduire à l’implantation d’un pace maker), (ii) parfois des troubles du rythme précoce (tachycardie/arythmie supraventriculaire ou ventriculaire), (iii) souvent une Cardiomyopathie Dilatée, (iv) plus rarement une atteinte musculaire (faiblesse musculaire, des membres inférieurs notamment). Un patient donné peut présenter une partie seulement des manifestations, et les manifestations peuvent varier d’un membre de la famille à un autre. Dans d’autres familles il s’agit de “Dystrophinopathies”, dues à des mutations du gène de la dystrophine. La maladie est transmise par le chromosome X, et elle peut s’associer à une myopathie. Parfois il n’y a aucune faiblesse musculaire mais uniquement une élévation d’un paramètre sanguin qui reflète une minime atteinte musculaire (CPK ou créatine phosphokinase).
Certains patients ne ressentent aucun symptôme (gène) dans leur vie quotidienne. Le diagnostic est alors fait à un stade précoce à l’occasion d’un examen médical fortuit ou motivé par une enquête familiale (devant un apparenté malade).
De nombreux patients ressentent des symptômes, essentiellement un essoufflement à l’effort (limitant la capacité d’exercice), plus rarement des palpitations (sensation inconfortable de percevoir ses battements cardiaques, irréguliers ou bien rapides), parfois des malaises, qui peuvent aller jusqu’à la perte de connaissance (habituellement en rapport avec une tachycardie rapide).
L’examen physique par le médecin peut être normal. Parfois le médecin retrouve des signes d’insuffisance cardiaque avec un bruit particulier à l’auscultation des poumons (des « crépitants »), un foie augmenté de volume, des oedemes des membres inférieurs (les chevilles en particulier).
La présence de symptômes doit alerter le patient. Celui-ci doit consulter son médecin pour faire un bilan cardiologique précis et déterminer le traitement adapté.
Le diagnostic est suspecté grâce à l’échographie cardiaque. Par l’utilisation des ultrasons, l’appareil visualise le cœur, permet de mesurer la taille des ventricules, analyse la contraction du muscle (via un paramètre appelé « fraction d’éjection » qui est ici abaissée), et étudie le flux sanguin (avec mesure du débit cardiaque) par un outil appelé Doppler.
 Figure 2
Figure 2
aspect en échocardiographie
Le diagnostic de CMD est ensuite établi quand le bilan ne retrouve pas de cause à cette dilatation / hypokinésie des ventricules. Ce bilan peut comporter une coronarographie (radiographie des artères coronaires) pour écarter le diagnostic d’infarctus, parfois un examen IRM, divers dosages sanguins.
Le bilan de la maladie consiste à caractériser le degré de sévérité, afin de déterminer précisément le traitement adéquat. Il fait appel à d’autres examens cardiologiques comme le Holter-ECG (enregistrement du rythme cardiaque pendant 24 heures), l’épreuve d’effort (exercice physique sur vélo ou tapis roulant) souvent couplé à une mesure de la consommation maximale d’oxygène (VO2max), des dosages sanguins (comme le taux de peptides natriurétiques : BNP ou NT-proBNP), parfois un cathétérisme cardiaque (mesure des pressions et débits par une sonde introduite dans le cœur par voie veineuse).
La maladie peut rester stable pendant une longue durée, ou bien évoluer vers une poussée d’insuffisance cardiaque avec essoufflement pour un effort minime voire au repos (c’est l’œdème pulmonaire aigu). La poussée est parfois favorisée par un écart au régime (un excès de sel souvent) ou une infection (une bronchite par exemple).
Des troubles du rythme cardiaque avec tachycardie (accélération excessive du cœur) peuvent survenir, et provenir soit des oreillettes (comme la fibrillation auriculaire, avec un risque de caillot sanguin et d’attaque cérébrale avec hémiplégie), soit des ventricules (comme la tachycardie/arythmie ventriculaire, avec un risque de perte de connaissance voire d’arrêt cardiaque et de mort subite).
Parfois un caillot sanguin se forme (appelé thrombus), qui peut migrer et donner selon les cas une embolie pulmonaire (migration dans une artère pulmonaire à partir d’une localisation veineuse) ou bien un accident vasculaire cérébral (migration dans une artère cérébrale à partir d’une localisation intra cardiaque).
La prescription de médicaments va permettre d’améliorer considérablement les symptômes. Les médicaments diurétiques permettent d’éliminer l’excès d’eau et de sel de l’organisme. Les inhibiteurs d’enzyme de conversion et les bêta-bloquants réduisent la surcharge de travail du cœur et améliorent son fonctionnement (les médicaments sont ici débutés à petite dose puis sont augmentés progressivement sur plusieurs mois). Les anti-coagulants (anti-vitamine K) sont parfois prescrits, pour fluidifier le sang et éviter la formation de caillot sanguin (le dosage est adapté à la mesure de l’INR sur des dosages sanguins réguliers). Des anti-arythmiques (amiodarone) sont parfois prescrits en cas de tachycardie ou troubles du rythme.
Des mesures hygiéno-diététiques sont souvent utiles, avec une consommation modérée de sel, un sevrage de l’alcool, un exercise physique modéré et régulier. Il existe par contre des restrictions vis à vis de certains sports avec exercise physique intense (voir la fiche spécifique du centre de référence sur l’activité sportive). Des programmes d’éducation thérapeutique et de ré-entrainement à l’activité physique peuvent être très utiles. La vaccination antigrippale est recommandée.
Les médicaments précédemment cités ont pour la plupart une action non seulement vis-à-vis des symptômes mais aussi une action préventive vis-à-vis des complications de la maladie.
Lorsque les médicaments s’avèrent insuffisants, d’autres stratégies thérapeutiques peuvent êtres discutées.
Chez les patients dont le bilan montre un risque important de complications rythmiques (troubles du rythme ventriculaire avec risque de mort subite), il peut être implanté préventivement sous la peau un appareil appelé défibrillateur automatique, qui reconnaît les épisodes de tachycardie grave et envoie un choc électrique permettant le retour en rythme cardiaque normal.
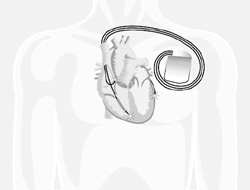
Le défibrillateur implantable
Le traitement de l’insuffisance cardiaque peut parfois conduire à l’implantation sous la peau d’un pace-maker avec une sonde dans chaque ventricule afin de les resynchroniser et d’améliorer ainsi la contraction du cœur et l’insuffisance cardiaque. Il s’agit ici d’un fonctionnement très particulier du pace maker appelé « resynchronisation » ou encore « stimulation biventriculaire ».
Dans des cas particuliers, et notamment en présence d’une cause rare de CMD (les laminopathies), un pace maker conventionnel peut être implanté pour détecter et palier aux épisodes de ralentissement excessif du cœur (bradycardie).
Enfin, dans les cas les plus sévères d’insuffisance cardiaque, certains patients peuvent bénéficier d’une transplantation cardiaque. La décision est prise conjointement avec un centre expert, qui continuera ensuite à suivre régulièrement le patient et adapter le traitement, notamment immunosuppresseur, pour éviter les épisodes de rejet de greffe.
La transplantation cardiaque
Les patients doivent être surveillés régulièrement en consultation avec leur cardiologue, et certains examensdoivent être refaits régulièrement (ECG, échographie cardiaque…) pour pouvoir adapter le traitement en fonction de l’évolution.
L’apparition de tout nouveau symptôme doit être signalé au cardiologue.
La grossesse chez une femme avec Cardiomyopathie Dilatée représente un risque médical important pour la maman (risque de complications, surtout en fin de grossesse et dans les mois qui suivent l’accouchement) qui conduit habituellement à déconseiller la grossesse. Il est recommandé de discuter à l’avance d’un projet de grossesse avec le cardiologue et le gynécologue pour aborder ces aspects médicaux.
Lorsqu’il s’agit d’une forme familiale de Cardiomyopathie, un bilan cardiologique (avec examen clinique, ECG, échographie cardiaque) doit être réalisé chez tous les apparentés au premier degré du patient (parents, frères et sœurs, enfants). Du fait de l’origine génétique et du mode de transmission habituellement autosomique dominant, la maladie peut en effet être transmise à la descendance avec un risque de 50% pour chaque enfant. Par ailleurs, la maladie a habituellement été transmise par l’un des deux parents du patient, et peut avoir été transmise à la fratrie de celui-ci. Chez l’enfant, le bilan doit être débuté au plus tard à l’âge de 10 ans, et il est souvent débuté plus tôt. Comme les signes cardiaques de la maladie sont souvent retardés, le bilan cardiaque doit être répété régulièrement (tous les 1 à 5 ans selon les cas), y compris chez l’adulte. Dans le cas particulier des Laminopathies, la surveillance doit comporter aussi un holter ECG, et les indications du pace maker et du défibrillateur doivent être précoces.
Le risque de transmission du gène anormal à la descendance justifie par ailleurs une consultation de conseil génétique en cas de projet de grossesse, de façon à discuter du risque de transmission, et des modalités de surveillance de l’enfant.
En l’absence de contexte familial de Cardiomyopathie (et en l’absence de mutation identifiée), une grande prudence est nécessaire. Un bilan cardiologique est habituellement préconisé chez les apparentés directs. S’il s’avère normal, il ne sera pas forcément renouvelé. Un conseil génétique spécialisé peut être utile.
Le test génétique consiste à faire une prise de sang, extraire l’ADN, et rechercher l’anomalie génétique (mutation) responsable de la maladie dans la famille. Le test peut être proposé chez le propositus (premier malade diagnostiqué dans la famille), surtout lorsqu’il s’agit d’une forme familiale de Cardiomyopathie (à discuter dans les autres cas). Actuellement une mutation est identifiée dans une faible proportion de cas (et au bout de plusieurs mois), sauf s’il y a un tableau clinique particulier et évocateur de laminopathie ou de dystrophinopathie (le taux de mutation est alors élevé).
La nature du gène et de la mutation va affirmer le mode de transmission, il peut parfois apporter au cardiologue une information complémentaire sur le risque évolutif de la maladie, et orienter la stratégie thérapeutique. Surtout, l’identification de la mutation chez un patient donné va permettre de proposer le test génétique chez ses apparentés, de façon à identifier les porteurs et non porteurs de mutation, et donc pouvoir guider la surveillance cardiologique au sein de la famille.
Coordonnées utiles :
- Le Centre National de Référence pour la prise en charge des maladies cardiaques héréditaires (Paris) : tel 01 42 16 13 47,
- Orphanet, site WEB de l’INSERM fournissant diverses informations médicales sur les maladies génétiques, et les consultations spécialisées,
- La Ligue contre la Cardiomyopathie (Association de patients atteints de Cardiomyopathies),
- 6 rue du Houssay, 28800 Montboissier ;
- [mail=ligue-cardiomyopathie@orange.fr]mail : ligue-cardiomyopathie@orange.fr[/mail] ;
- site web : http://www.ligue-cardiomyopathie.com
- L’association des patients porteurs de défibrillateur
Documents joints
Fiche sports
Recommandations pour la participation aux activités sportives de loisirs pour les patients atteints de Cardiomyopathie Dilatée
Le sport en compétition est formellement interdit.
Les recommandations qui suivent concernent l’activité de loisir et sont données à titre indicatif et doivent être adaptées au cas par cas (selon les symptômes, les résultats du bilan cardiologique précis, l’effet contrôlé du traitement médicamenteux), en accord avec le cardiologue du patient.
D’une façon générale les principes suivants peuvent être proposés :
D’une façon générale les principes suivants peuvent être proposés :
- Eviter une activité sportive avec épisodes répétés d’accélération/décélération (exemple tennis, basket, football). Il faut favoriser au contraire les sports avec activité progressive ou stable.
- Eviter des conditions environnementales extrêmes : froid intense, chaleur intense, forte humidité, haute altitude.
- Eviter les programme visant à atteindre et repousser ses limites. Il faut au contraire rester en deçà d’une limite physique adaptée à la maladie.
- Eviter toute activité physique qui déclenche les symptômes de la maladie.
- Eviter les activités foraines ou parc de jeux associés à une forte émotion, des accélérations soudaines de la fréquence cardiaque…
- Eviter les activités à risque en cas de malaise ou de perte de connaissance, telles que alpinisme, plongée sous marine.
- Eviter tout produit visant à améliorer les performances physiques (y compris des compléments nutritionnels de type « ma huang » qui comporte un stimulant cardiaque aux effets délétères potentiels).
- Les patients avec implantation de défibrillateurs ne sont pas libérés des restrictions sportives proposées ici. Les conseils rapportés ici s’appliquent également à eux. Certaines activités sont de plus à éviter car pouvant endommager le matériel ou déclencher un choc électrique inapproprié.
Les recommandations suivantes sont regroupées en 3 catégories :
- activités sportives contre indiquées ou déconseillées (Proscrire) ;
- activités à évaluer individuellement (A évaluer) ;
- activités probablement autorisées (Autoriser).
| Intensité élevée | Recommandations chez patient avec CMD |
|---|---|
| Basket | Proscrire |
| Body Building | Proscrire |
| Hockey sur glace | Proscrire |
| Squash | Proscrire |
| Escalade | Proscrire |
| Course à pieds (sprint) | Proscrire |
| Ski | A évaluer |
| Football | Proscrire |
| Rugby | Proscrire |
| Tennis (simple) | Proscrire |
| Planche à voile | Proscrire |
| Intensité moyenne | Recommandations chez patient avec CMD |
|---|---|
| Baseball | A évaluer |
| Vélo | Autoriser |
| Jogging | A évaluer |
| Surf | A évaluer |
| Voile | A évaluer |
| Natation | Autoriser |
| Vélo d’appartement | Autoriser |
| Haltérophilie | Proscrire |
| Randonnée soutenue | A évaluer |
| Tennis (double) | A évaluer |
| Randonnée tranquille | Autoriser |
| Intensité faible | Recommandations chez patient avec CMD |
|---|---|
| Bowling | Autoriser |
| Golf | Autoriser |
| Equitation | A évaluer |
| Plongée sous-marine | Proscrire |
| Patinage | Autoriser |
| Nage avec masque | Autoriser |
| Marche rapide | Autoriser |
Inspiré de Maron et al, Circulation 2004 ; 109 : 2807-2816.